mamba create -y –n entrez entrez8 Sequence analysis
In this chapter we we will consider several biological concepts that appear central to understand the manipulation of biological data.
We are also covering the transition of a biological (organic) information to digital bits.
We will explore some databases in which biological information is stored.
8.1 Biological information
Since the origin, organisms (or molecules) have been the result of different selective processes. An emergent property of successive iterations of survival/decease was the ability of molecules to keep a record of its past in a very stable manner, so that it will pass generation to generation. Although this might not be the first property or molecule to ever exist (see the :origin of life), the innovation of organisms to pack information of previous events became so advantageous that almost any form of living organism has this property: that is DNA.
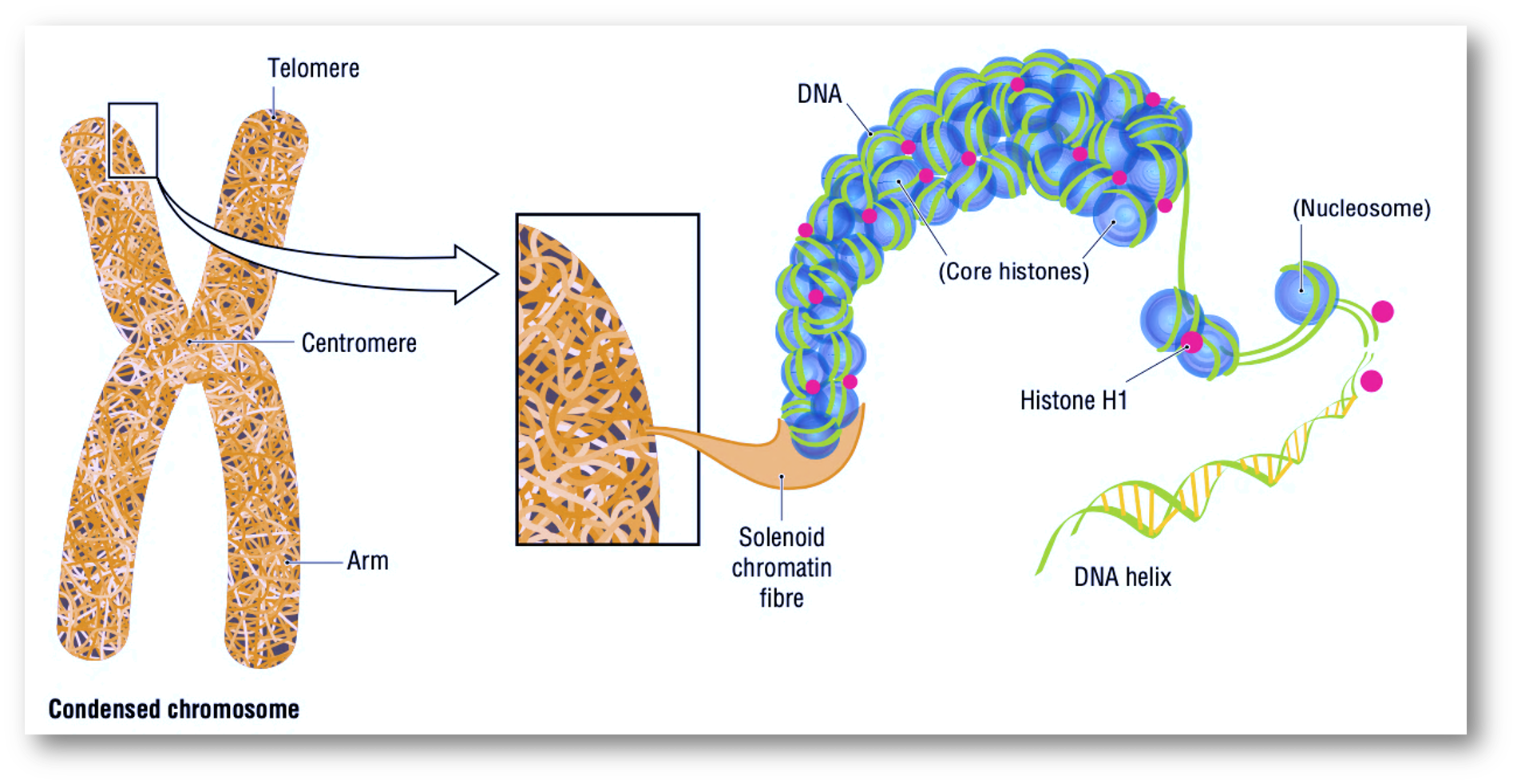
Almost any living organism harbor DNA in a nucleus or chromosome, which is a very compacted structure that could be actually seen to naked eye in some species. It so arranged or condensed Fig. 8.1 that it is the result of multiple compacting strategies.
8.2 The duality of DNA
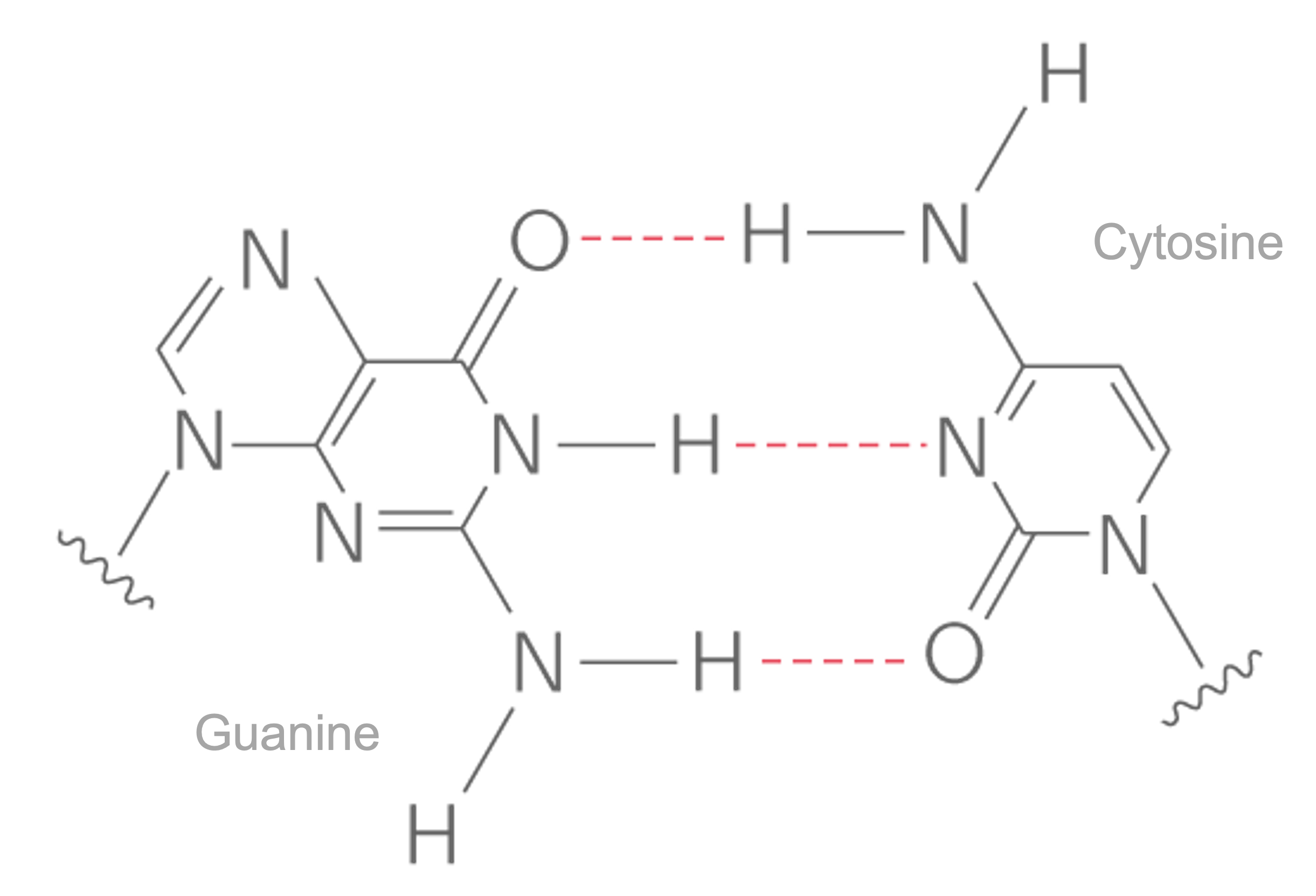
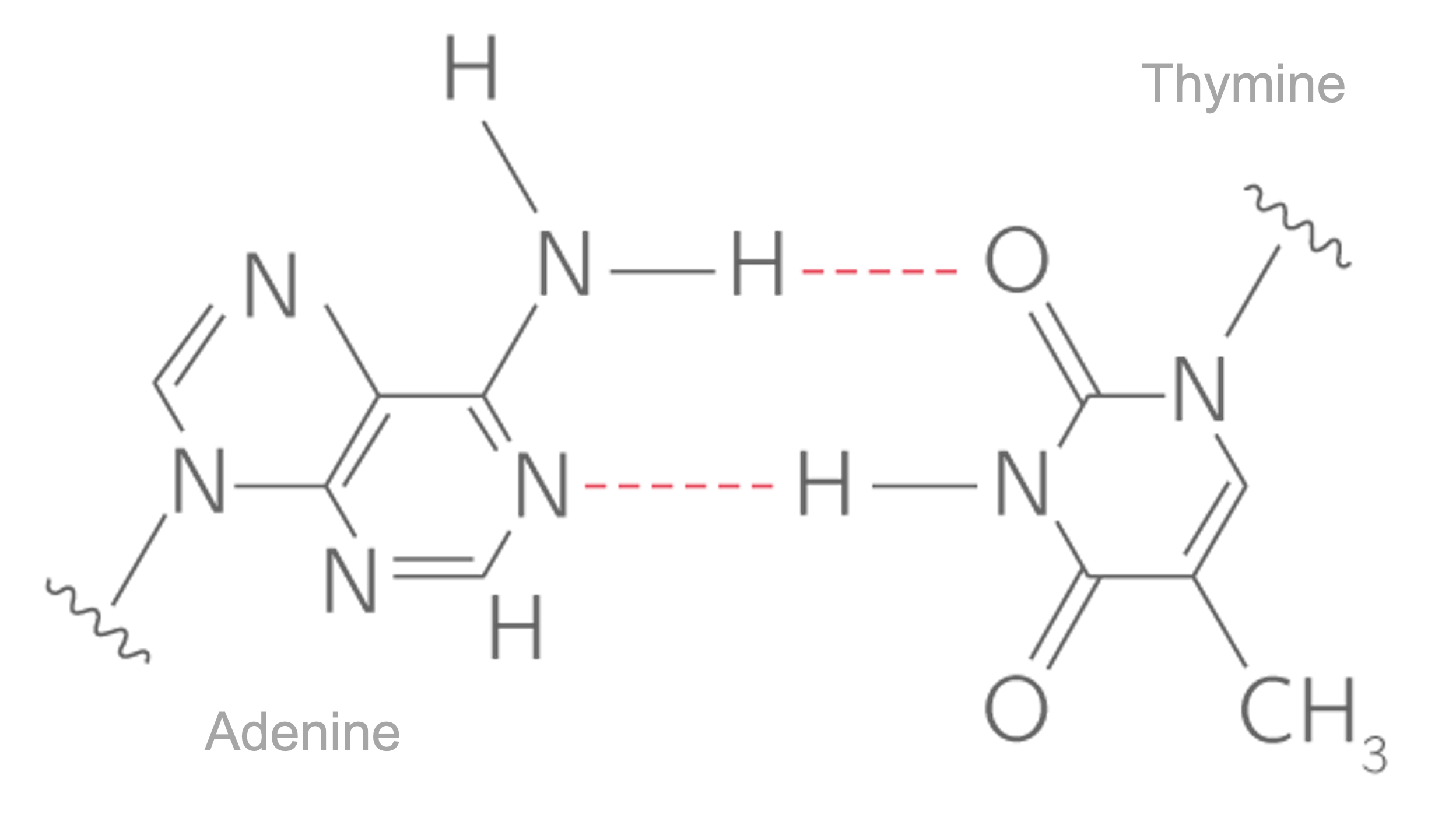
DNA is an organic molecule of :nucleic acids that is mainly build out of nitrogen-containing compounds (:nucleobases) or just bases.The four bases are adenine (A), cytosine (C), guanine (G) and (T) and they bind strictly as \(A=T\) and \(C\equiv{G}\) where each of the lines represents an hydrogen bond.
DNA nitrogenous bases of are arranged in a very compacted helix structure and too much can be understood from its molecular nature. It is of course governed by the physicochemical properties of the atoms, therefore DNA is a physicochemical entity. But those common bricks have also a very beautiful emergent property that comes out from their order (mainly), and that is to keep the instructions that build the organism from which it belongs. These instructions are nothing else than information, which also follow some informational rules. Then DNA is also an informational entity.
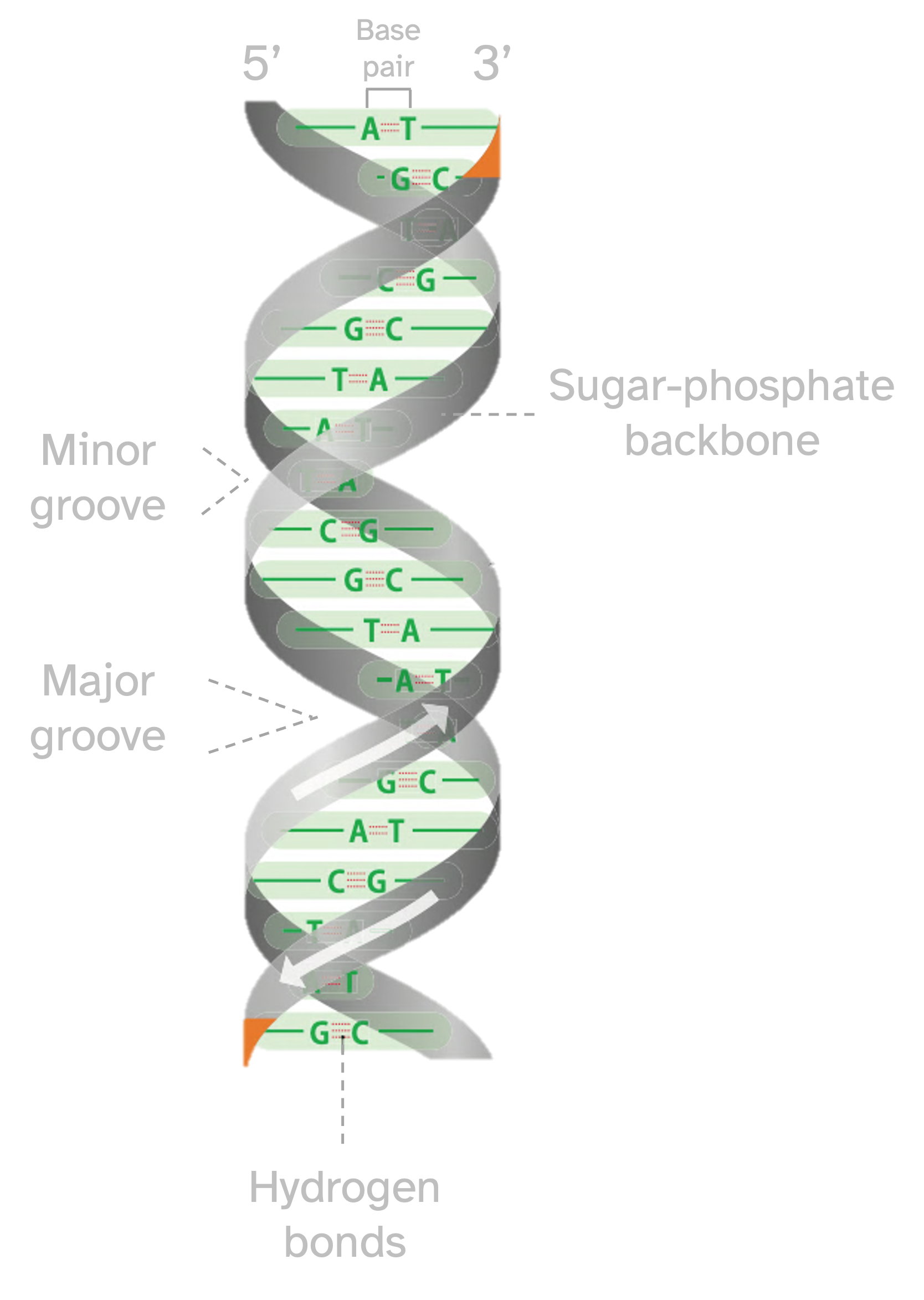
The description of DNA bases helped the elucidation of the helicoidal structure as well as the elucidation of the base pairing rules of the nucleotides Fig. 8.4, also called the Chargaff’s rules of base pairing. The first outstanding feature of the DNA structure was indeed highlighted by Watson and Crick in its famous paper about a subtle mechanism of replication. The
This idea of the duality of the DNA (and of course of other biological molecules as well) is the source of the study of many bioinformatic fields. But a question remains open: how do we capture the informational nature of a DNA sequence so that we end up with a sequential file of characters ATCGCTATC.... This is not a trivial question in fact.
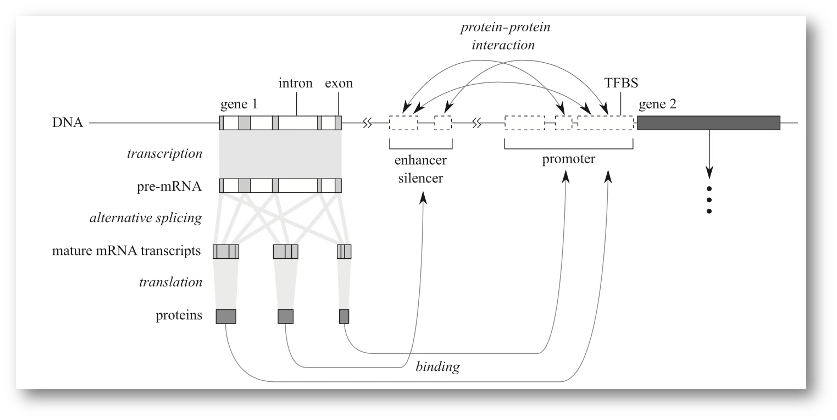
8.3 The central dogma theory of molecular biology extended
So far we have addressed that DNA is a very stable molecule that stores biological (i.e., evolutionary) information. Also, that DNA could represent a sequential object of characters as in a computer digital object or file as well. But how is it that this order has an underlying biological sense? This was a very though question that required the accumulation of many experimental discoveries and the meeting of genetics and molecular biology.
8.4 Sequencing strategies
8.5 Sequencing over time
8.6 Some insights from sequencing genomes
8.7 Biological information databases
8.8 Retrieving data from NCBI
Inforation or databases inside the NCBI comes in many different ways. Perhaps the most used file format in bioinformatics is the FASTA format. It is a very simple format that consist of two lines, the header which is basically noted by starting with the > symbol and is always preceding the sequence, that is present in the second line.
8.8.1 The manual way
8.8.2 Entrez direct
We first need to install the Entrez Direct command line utilities. This could be done in several ways, one suggested by the manual and a very simple one using conda (mamba).
This line will create a new conda environment called entrez and will automatically install all dependencies. We later activate the environment with conda activate entrez and then we got the entire Entrez CLI utilities.
There are several commands and its function is to interact with the NCBI databases system. It is, however, a very huge set of utilities and commands to cope with at first, so the learning curve is somehow steppe. Here is a way to download the Bacillus tequilensis EA-CB0015 genome with a line of code that interleave three of the main commands of Entrez:
esearch -db assembly -query GCF_012225885.1 |
elink -target nucleotide -name assembly_nuccore_refseq |
efetch -format fasta > GCF_012225885.1.fasta8.8.3 Simpler download programs
Since the manual way is clumsy when scaling and automating, and the entrez-direct utilities are somehow hard at first hand, some bioinformaticians have already thought ways to ease users the hard of knowing the NCBI internal database structure and to resume many of the utlities to just a ‘download’ command out from what in entrez is distributed in search, filter, and download commands.
A very nice command to download genomes is the bit-dl-ncbi-assemblies from Mike Lee, who has also made a remarkable work to bring many bioiformatic knowledge to newcomers. Similarly and quite more robust is the ncbi-genome-download CLI (or abbreviated as ngd) developed by Kai Blin
When navigating the NCBI databases some data is being curated and continuously updated by the execution of annotation tools. The refseq database is precisely the one that is more curated. For the case of genomes the accession number of those that belong to the refseq category are start with GCF ids, whereas the annotated genomes from the genbank category display the GCA ids
Using ngd to download a complete genome using the refeseq or genbank accession numbers, would be as easy as:
ngd --format fasta --flat-output -A GCF_002055965.1 bacteriaThe one-line will download the refeseq genome of the Bacillus tequilenis EA-CB0015 in a fasta nucleotide format. the --flat-option from the script will handle out extra metadata of the search and will download strictly the data.
It is important to highlight that these simpler scripts are focused on genomic information, so that direct downloads of genes, proteins, transcripts, and many other kind of data must be done with entrez or other programs. Some recent efforts have been made to create a more robust CLI to interact with different databases, but in a very easy and intuitive way, such as the gget project. It is more focused on vertebrate databases, but also has a port to NCBI, UniProt and other databases. And is very simple. Here is an example to download all the isoforms in a multi-fasta format of the cancer-related gene BRCA2 in a human genome (GRCh38.p13):
gget seq -iso -o BRCA2.fasta ENSG00000139618It is establishing a connection with the Ensemble database, which is focused on vertebrate genome and transcriptome data.
Challenge
Professor Camilo is interested on knowing how many complete genomes of Bacillus subtilis are there in the NCBI databases. He asks you later to count the number of features (genes, CDS, ncRNA, rRNA, etc.) in the genome of Bacillus subtilis NCIB 3610 (GCF_002055965.1). And tell you to document each of the steps and how did you end up with the answer. Saving the file with your initials (e.g., CG-activity01.md)